ADLab at Indian Association for the Cultivation of Science, Kolkata (IACS)
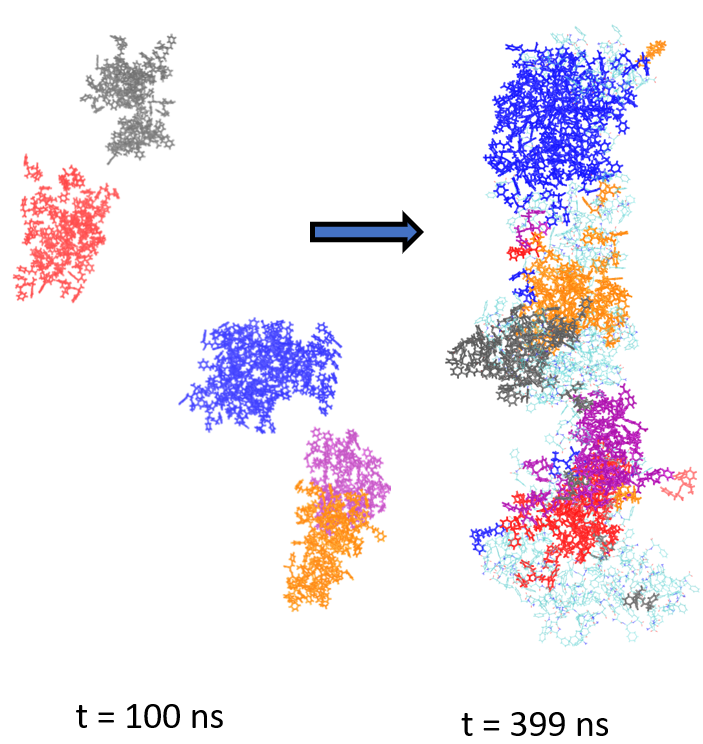
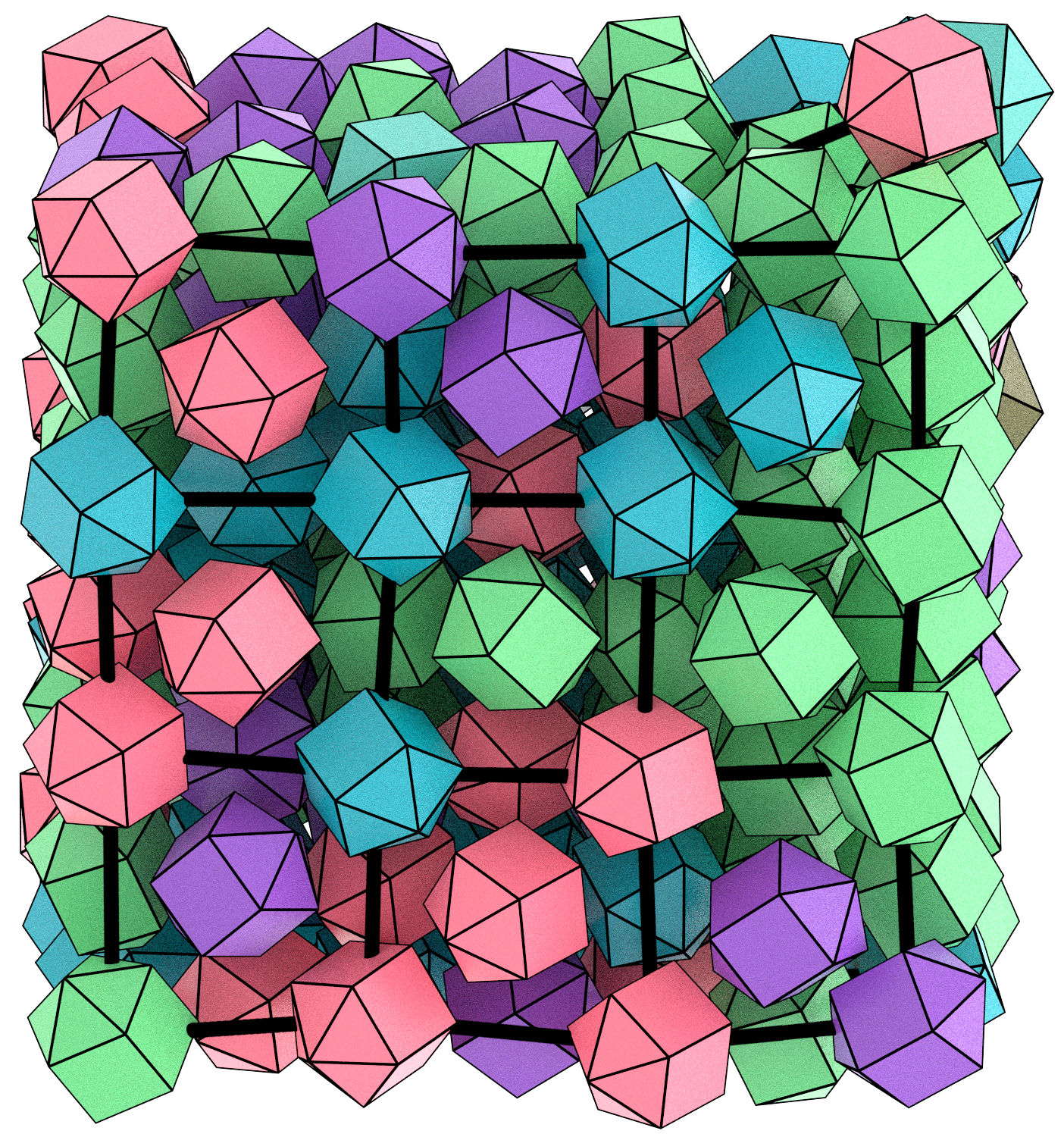
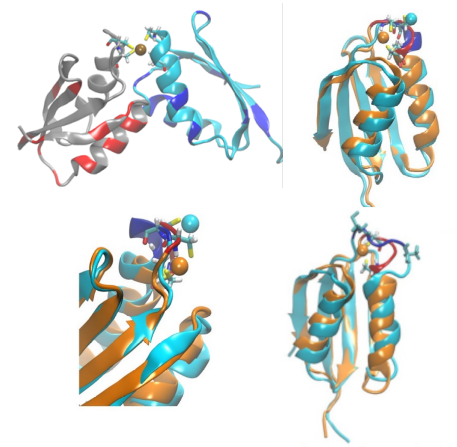
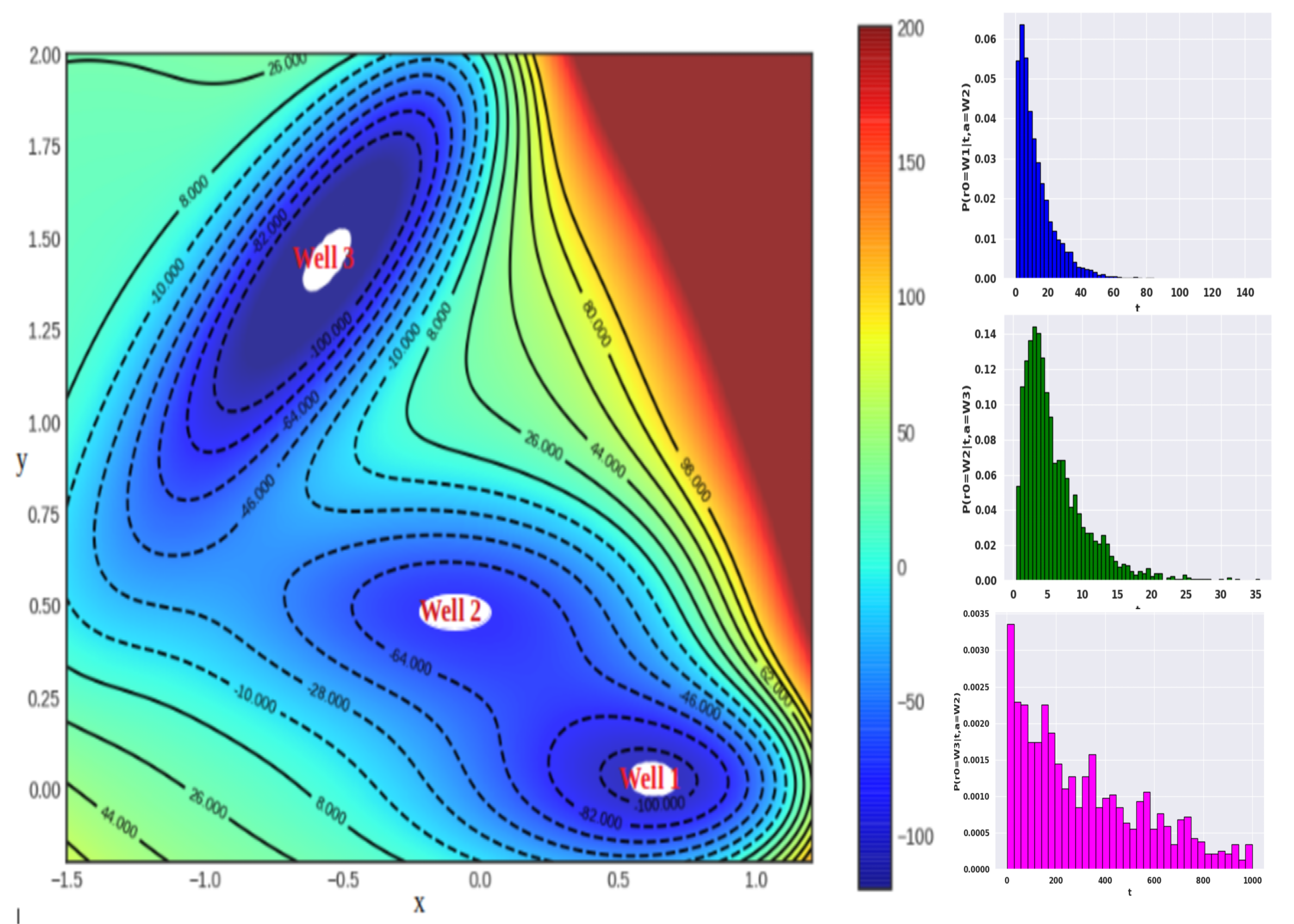
We are a theoretical and computational group interested in understanding structure and dynamics of complex systems using classical statistical mechanics and electronic structure methods. Our research program has an equal emphasis on development of new simulation methods and application of existing/new methods to important problems in chemistry, molecular biophysics, bioinorganic chemistry and soft matter systems.